Trajectory inference
Asc-Seurat supports three trajectory methods that share the same Shiny controls:
Slingshot (default) — fits lineages on the clustered UMAP embedding. Start and end clusters can guide lineage orientation, and the results include a compact minimum-spanning-tree graph of the inferred cluster relationships.
PAGA — estimates connectivity between clusters and calculates pseudotime from the selected root cluster.
Monocle 3 — learns a Monocle 3 principal graph on the existing UMAP and orders cells from the selected root cluster.
Running trajectory inference
The Trajectory Inference tab takes a clustered Seurat object as input. You can load it in two ways:
Upload from browser — the default and most convenient option for moderate-sized objects.
Path on this computer/server — best for very large RDS files, where the browser upload limit is a concern. Relative paths are resolved against the working directory shown beneath the input.
For all three methods you can:
pick the trajectory method (Slingshot / PAGA / Monocle 3),
choose a root/start cluster; this is strongly recommended so the pseudotime orientation is explicit,
optionally fix an end cluster (Slingshot only),
inspect the resulting pseudotime plot and trajectory graph,
export the trajectory object.
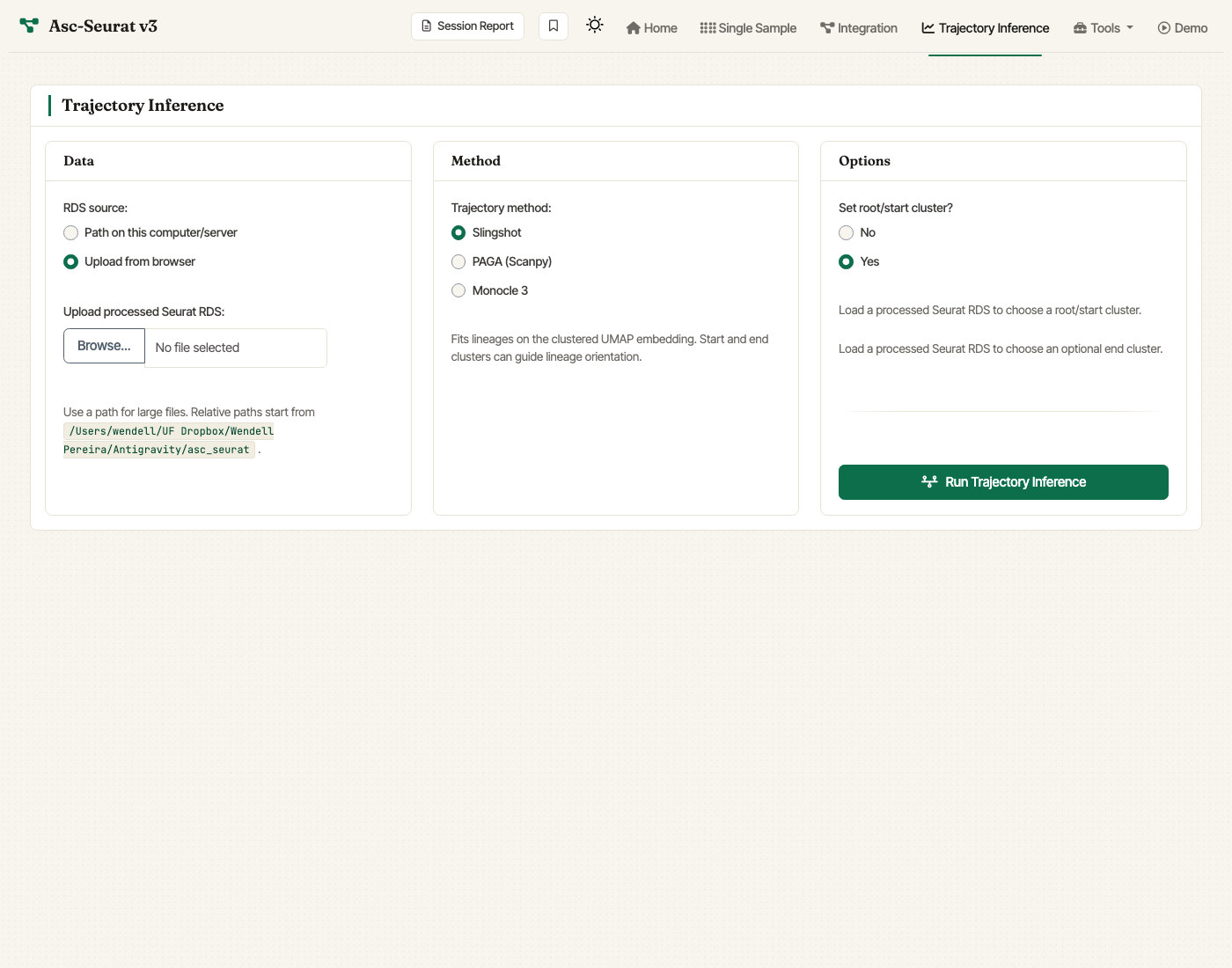
Trajectory Inference tab. Upload a clustered Seurat RDS or provide a server path, pick Slingshot, PAGA, or Monocle 3, and set a root/start cluster before running inference.
Trajectory gene discovery
Each trajectory method exposes a gene-discovery workflow that matches the method’s underlying model.
For Slingshot, the Shiny workflow exposes three trajectory-DE engines:
PseudotimeDE-fast (recommended default). Fast, well-suited to the Slingshot pseudotime produced by Asc-Seurat.
scMaSigPro. Polynomial-regression alternative; identifies genes that change significantly along pseudotime.
tradeSeq. More flexible but slower; the UI labels it as a long-runtime option and lets you tune the number of knots.
Set the Top genes to display/plot control to limit how many top genes are shown in the displayed table and plots; all variable genes are tested regardless.
For PAGA, the workflow focuses on connected states rather than lineage-specific DE:
Connected cluster markers compares selected or top-connected PAGA cluster pairs with Seurat marker testing.
Pseudotime-associated genes ranks genes that vary along cells in the selected connected clusters.
For Monocle 3, Asc-Seurat uses Monocle 3’s graph-based test to rank genes that vary over the learned principal graph. The optional gene-module step groups significant trajectory-variable genes into modules.
Installing the Python and R dependencies
The Docker image bundles the trajectory and DE dependencies. The R package install also installs the required R trajectory packages; only the Python stack for PAGA is prepared separately:
PAGA: requires
scanpy. Runascseurat::setup_paga()from R to verify or prepare the Python environment.Monocle 3, PseudotimeDE-fast, scMaSigPro, and tradeSeq are declared as app runtime dependencies.