Loading the data and integration of multiple samples
To analyze multiple samples, select the Integration tab in the web application. The full integration workflow is contained in this tab: sample declaration, per-sample QC, normalization, integration method, clustering, and the integrated visualization handoff.
Integration uses Seurat v5’s IntegrateLayers framework. RPCA is the default method and Harmony is also available from the same tab. Samples are declared directly in the UI — pick the number of samples, type their names and paths inline, and set the QC values for each on the same page.
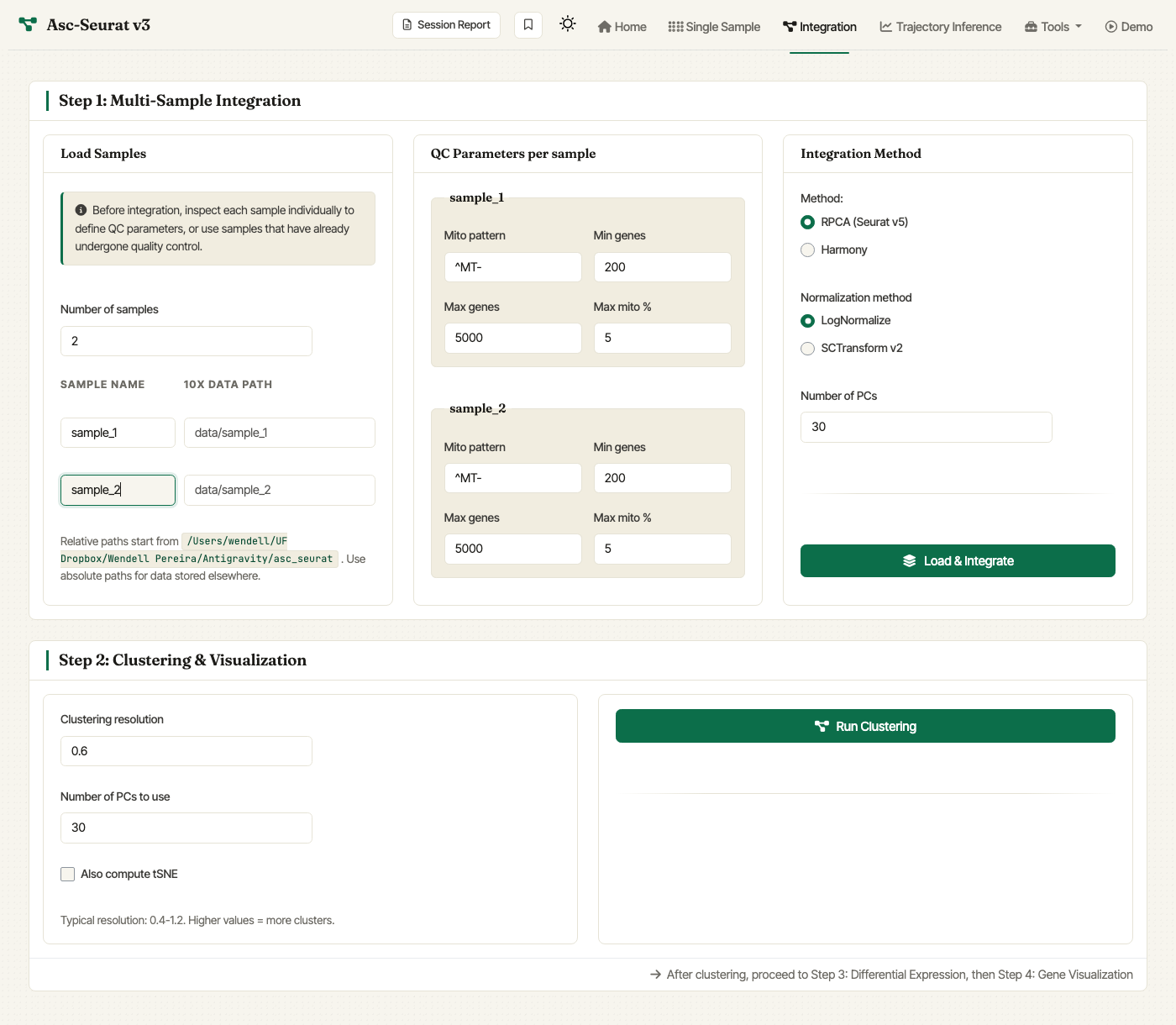
Integration tab. Declare the number of samples and their paths in Load Samples, set per-sample QC in the middle column, and pick RPCA (Seurat v5) or Harmony in Integration Method.
Declaring the samples
In the Load Samples card:
Set Number of samples (1–24).
For each sample, fill in:
Sample name — any label you prefer. Use the same name for biological replicates that should be pooled in plots and analyses.
10X data path — relative or absolute path to the sample’s directory.
Setting per-sample QC parameters
In the QC Parameters per sample card, for each declared sample set:
Mito pattern — regular expression matching mitochondrial gene IDs (default
^MT-for human; use^mt-for mouse,^ATMGfor Arabidopsis, etc.).Min genes — minimum number of genes a cell must express to be retained.
Max genes — maximum number of genes a cell can express (useful to filter suspected doublets).
Max mito % — maximum percentage of mitochondrial transcripts allowed per cell.
Choosing the integration method
In the Integration Method card:
Method:
RPCA (Seurat v5)(default) orHarmony.Normalization method:
LogNormalize (faster)(default) orSCTransform v2.Number of PCs: principal components to use during integration (default
30; range 5–100).
Click Load & Integrate to run the integration. The integrated object becomes available to the clustering, DE, and visualization modules below.
Saving the integrated object for reuse
Integration is the most time-consuming step in this tab. After it completes, an Integration Summary card lets you download the integrated Seurat object as an RDS file. To skip the integration step later, load that RDS in the Single Sample tab using Existing Seurat RDS — the clustering and downstream steps will pick up from there.